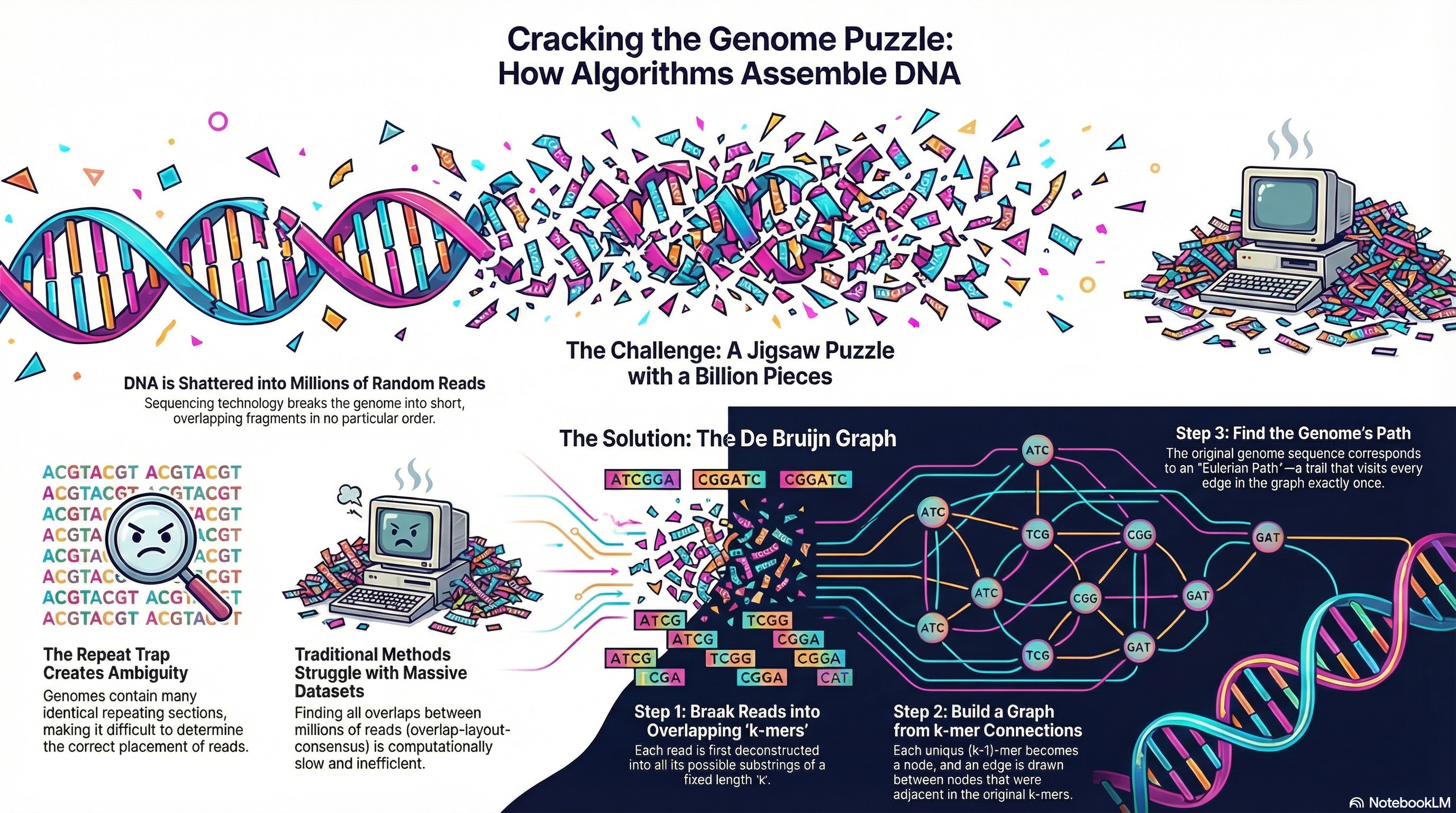
基因组组装(Genome Assembly)在本质上是一个逆问题:我们试图从海量有噪声的观测数据(Reads)中,还原出唯一的、连续的原始信号(Genome)。
问题定义
我们首先需要定义问题。假设存在一个未知的原始基因组序列 ,其长度为 。测序过程可以被视为一个随机采样函数,它从 中生成了 个子串(Reads),每个 Reads 的长度为 ,且 。理想状态下,这些 Reads 覆盖了基因组的每一个碱基,且拥有足够的冗余度(Coverage)。我们的任务是寻找一个函数 ,使得 。
这里的核心矛盾在于“重复”。如果基因组 是一个完全随机的序列,不包含任何重复子结构,那么组装问题仅仅是一个简单的排序问题。然而,生物学基因组充满了转座子、串联重复和片段复制。当测序读长 小于重复片段的长度 时(),单一的 Read 无法跨越重复区域,这就导致了局部信息的模糊性。在图论中,这种模糊性直接转化为路径的多样性,即无法唯一确定欧拉路径或哈密顿路径。
OLC 算法
当我们拿到一堆碎片时,人类最本能的反应是寻找碎片边缘的相似性。如果碎片 A 的尾部与碎片 B 的头部完全一致,我们就有理由相信 A 位于 B 之前。这就是 Overlap-Layout-Consensus (OLC) 算法的核心直觉。
图的构建:以 Reads 为节点
在 OLC 模型中,我们构建一个有向图 。
每一个节点 代表一条完整的 Read。
每一条边 代表 Read 的后缀与 Read 的前缀存在显著的重叠(Overlap)。
哈密顿路径
构建好重叠图后,组装问题就转化为:在图中寻找一条路径,该路径经过图中的每一个节点恰好一次。这在图论中被称为 哈密顿路径(Hamiltonian Path) 问题。
NP-hard
虽然 OLC 模型完美地保留了 Read 内部的连贯性(Coherence),但它在数学上是昂贵的。寻找哈密顿路径是一个著名的 NP-hard 问题,意味着随着节点数量的增加,计算时间呈指数级增长。
更为致命的是图构建阶段的计算量。为了建立边,我们需要将每一条 Read 与其他所有 Reads 进行比对。这是一个 复杂度的操作。
内存瓶颈
在二代测序(NGS)时代,为了获得高覆盖度,我们通常会产生数以亿计的短 Reads()。如果我们试图构建一个 的重叠矩阵或邻接表,所需的内存将是天文数字。例如,对于人类基因组,即使仅存储稀疏图,内存消耗也往往超过 TB 级别,且计算两两比对的时间在单机上可能需要数年。
因此,OLC 算法虽然直观且能保留长距离连接信息,但在处理海量短读长数据时,它撞上了计算复杂度的物理墙。我们需要一种新的数据结构,将问题从 降维。
德布罗因图(De Bruijn Graph)
我们不再试图保留整条 Read,而是将其打碎,将“比对”问题转化为“查表”问题。
k-mer 化
我们将每条 Read 切割成长度为 的短片段,称为 k-mer。
例如,若 Read 为 ATGCG,k=3,则产生的 k-mers 为 ATG, TGC, GCG。
这一步看似是“幼稚”的(childish),因为它丢失了 Read 内部原本存在的长距离连接信息(Read Coherence),例如 ATG 和 GCG 原本确实在同一条 Read 上,但在切碎后,这种物理连接被切断了。但正是这种“破坏”,换来了计算的可行性。
图的构建:以边为中心
在 DBG 中,定义的规则发生了翻转:
节点(Node):是长度为 的序列。
边(Edge):是长度为 的 k-mer。
对于一个 k-mer ATGC,它对应一条从节点 ATG 指向节点 TGC 的有向边。
算法目标:欧拉路径
现在,组装问题转化为:在图中寻找一条路径,该路径经过图中的每一条边(即每一个 k-mer)恰好一次。这就是 欧拉路径(Eulerian Path) 问题。
与哈密顿路径不同,寻找欧拉路径存在线性时间 的算法。只要图是连通的且满足节点的度数平衡(入度等于出度),我们就一定能找到解。
内存消耗
在 DBG 中,内存消耗不再取决于 Reads 的数量(测序深度),而是取决于 基因组的大小和复杂性(即图中唯一的 k-mer 数量)。无论测序深度是 50x 还是 500x,对于同一个物种,其 k-mer 种类数是相对固定的(受限于基因组大小)。通过使用哈希表(Hash Table)或布隆过滤器(Bloom Filter)存储 k-mer,我们成功地将内存消耗控制在了可接受的范围内(例如人类基因组组装通常需要 100GB-500GB 内存)。
DBG并不是完美的替代
虽然 DBG 解决了计算效率问题,但它引入了新的麻烦:由于我们将 Reads 切碎了,所有的重复序列在图中不再保留其原始的位置信息,而是物理地合并在了一起。这就是你提到的“图拓扑结构中的坍缩”。为了直观理解这个概念,我们构建一个最小化模型。
重复导致的节点坍缩
假设我们真实的基因组片段包含两个区域,中间共享一段重复序列 R。
序列 1:L1 - R - R1 (例如:AAA - TGT - CCC)
序列 2:L2 - R - R2 (例如:GGG - TGT - TTT)
这里,TGT 是重复序列,长度为 3。
如果我们设定 k-mer 大小 (即节点长度 ):
-
处理序列 1:生成路径
AA→AT→TG→GT→TC→CC。 -
处理序列 2:生成路径
GG→GT→TG→GT→TT→TT。
请注意中间的 TG 和 GT 节点。
在序列 1 中,TG 来源于 AAA-TGT-CCC 的左侧上下文。
在序列 2 中,TG 来源于 GGG-TGT-TTT 的左侧上下文。
但在 DBG 的哈希表中,TG 只是一个键值。因此,来自序列 1 和序列 2 的 TG 节点被强制合并为一个物理节点。
拓扑结构的结果:
在图中,我们会看到一个形状像“X”或者是“缠结(Tangle)”的结构。
-
节点
TG拥有两个入度(In-degree):分别来自AT(序列1) 和GT(序列2,甚至可能形成环)。 -
节点
GT拥有两个出度(Out-degree):分别指向TC(序列1) 和TT(序列2)。
当组装算法遍历到这个“坍缩”的重复节点时,它面临歧义:应该走哪条路?数学上,这意味着存在多条可能的欧拉路径。我们丢失了“谁原本和谁连在一起”的信息。这就是 DBG 将长 Read 切碎所付出的代价:特异性(Specificity)的丧失。
利用双端测序(Paired-End)
既然 DBG 本身无法区分重复节点的出口,我们需要引入外部信息。这就是 Mate Pairs 或 Paired-End Reads 发挥作用的地方。
虽然单独的 k-mer 很短,但我们知道两个 k-mer 是一对,且它们在基因组上的物理距离(Insert Size)是固定的(例如 500bp)。
在上述模型中,虽然算法在重复节点 R 处迷路了,但我们有一对 Read:一端位于 L1,另一端位于 R1。
-
Read Pair: (AAA…, …CCC)
这个配对信息就像一座“高架桥”,跨过了混乱的 R 节点,告诉算法:如果你是从 AAA 进来的,你就必须去 CCC 那边,而不能去 TTT 那边。
在实际组装器(如 SPAdes 或 Velvet)中,这一步通常发生在图构建之后。算法首先简化图(将无歧义的路径压缩为 Contigs),然后利用 Read Pairs 将这些 Contigs 定向连接起来,形成 Scaffolds,从而解开(Untangle)由于重复序列导致的拓扑死结。
长读长时代的 OLC 复兴
在很长一段时间里,基因组学被困在“短读长(Short Reads)”的微观视野中。虽然 Illumina 测序提供了极高的准确度(99.9%),但其 150bp 的读长在面对人类基因组中成千上万碱基的转座子(如 LINEs, SINEs)时显得无能为力。这就好比试图用指甲盖大小的碎片去拼凑一幅巨大的蓝天拼图,每一片看起来都一样,位置无从从确定。
打破 的诅咒
第三代测序技术(PacBio 和 Oxford Nanopore)的出现,从物理层面改变了组装问题的数学性质。当读长 显著增加,直到超过基因组中最长重复序列的长度 时(即 ),原本在德布罗因图(DBG)中形成的复杂“缠结”和“气泡”会瞬间解开。
从图论的角度看,如果一条 Read 能够完整覆盖整个重复区域及其两端的唯一序列,那么在重叠图(Overlap Graph)中,这个重复区域就不再是一个歧义的汇聚点,而是一个可以被确定的局部路径。这使得我们不再需要为了计算效率将 Read 切碎成 k-mer,因为 Read 本身已经包含了最重要的连接信息。于是,古老的 OLC(Overlap-Layout-Consensus) 算法逻辑重新成为了主流。
早期长读长和含噪声OLC
早期的长读长技术(如 PacBio CLR 和早期的 Nanopore)虽然读长达到了 10kb-100kb,但伴随着极高的错误率(约 10%-15%)。这种高噪声数据让传统的 OLC 算法面临挑战:当两条 Read 的序列相似度只有 85% 时,如何判断它们是真正的重叠,还是源自不同的基因组区域?
为了解决这个问题,诞生了 含噪 OLC(Noisy OLC) 策略,典型代表是 Canu 和 FALCON。
这些算法引入了概率模型来处理噪声:
校正(Correction): 并非直接进行组装,而是利用 Read 之间的相互重叠进行“自我纠错”。通过堆叠多条含噪 Read,高频出现的碱基被认为是真实的信号,而随机分布的错误被过滤掉。
修整(Trimming): 切除低质量的末端。
组装(Assembly): 在校正后的 Read 上构建重叠图。此时使用的是 String Graph(串图),这是 OLC 的一个变体。与朴素的 OLC 图不同,String Graph 进行了传递归约(Transitive Reduction)。
String Graph 的传递归约
如果存在重叠关系 , ,以及 ,那么边 是冗余的(Transitive Edge)。String Graph 会删除这条边,只保留最长路径。这一步极大地简化了图的结构,降低了内存消耗,使得处理大基因组成为可能。
HiFi 长读长测序
真正的转折点发生在 PacBio HiFi(High Fidelity) 测序技术的成熟。HiFi 模式通过对同一个 DNA 分子进行多次环形测序(CCS),在保持 15kb-20kb 长读长的同时,将准确率提升到了惊人的 99.9%。
这是一种“双重红利”:我们既拥有了跨越重复序列的长读长,又拥有了无需复杂纠错的高准确度。这直接催生了新一代组装算法,如 hifiasm。
Hifiasm 采用了一种更加激进且优雅的策略。它不再仅仅满足于组装出一致性序列(Consensus),而是致力于 单倍型分型(Haplotype Phasing)。人类是二倍体生物,拥有父本和母本两套染色体。传统的组装往往将两者混合,生成一个“嵌合”的基因组。而 Hifiasm 利用 HiFi 数据的高准确性,能够区分父本和母本之间微小的单核苷酸多态性(SNP),从而在组装图中将两条单倍型路径彻底分开,生成 Phased Assembly。此时的组装图不再是一条混合的线,而是像拉链解开一样,清晰地展示出两条平行的染色体路径。
T2T 组装与着丝粒
即便有了 HiFi,基因组中着丝粒(Centromeres)和端粒(Telomeres)仍然是一个问题。这些区域由数百万碱基的超长串联重复序列(Satellite DNA)组成,其重复单元极其相似,且总长度往往超过 HiFi 的 20kb 读长。在这些区域,HiFi 组装图依然会断开。
这就是 端粒到端粒(T2T) 组装的目标:不留任何缺口(Gap),完美还原每一条染色体从头到尾的序列。人类基因组 T2T-CHM13 参考序列的发布标志着这一时代的到来。
T2T 的组装策略:Verkko 算法
为了攻克着丝粒,我们需要更长的武器——超长纳米孔测序(ONT Ultra-Long Reads),其读长可达 100kb 甚至 1Mb+。但 ONT 数据的错误率相对较高。因此,最新的 T2T 组装器如 Verkko 采用了一种混合图策略:
-
HiFi 构建骨架: 首先利用高准确度的 HiFi Reads 构建一个非常坚固但不连续的 德布罗因图(DBG)。在这个图上,非重复区域形成了完美的线性路径,而着丝粒区域则形成了复杂的“纠缠结”。
-
ONT 穿针引线: 将超长 ONT Reads 映射到这个 HiFi DBG 上。由于 ONT Read 足够长,它们可以像一条条线索,穿过那些混乱的重复节点,指出正确的出口方向。
-
解结与连接: 算法利用 ONT 的跨越信息,解开 HiFi 图中的复杂节点,将断开的 Contigs 连接成完整的染色体 Scaffolds。
这种结合了“HiFi 的精确图结构”与“ONT 的超长路径约束”的策略,是目前实现 T2T 组装的标准范式。
评价指标
当我们完成组装后,如何评价其质量?生物信息学中存在一套基于统计学和生物学的评价体系。
N50 是最常被引用的指标,但也是最容易被误解的。它的定义是:将所有 Contigs 按长度从大到小排序,累加长度,当累加值达到总基因组长度的 50% 时,当前这个 Contig 的长度就是 N50。
N50 不代表准确性
N50 仅仅衡量了组装的 连续性(Contiguity)。一个充满错误拼接(Mis-assembly)的组装结果可能拥有极高的 N50(因为强行把不相关的片段连在了一起),但它在生物学上是毫无价值的。因此,N50 必须结合准确性指标一起看。
为了弥补 N50 的缺陷,我们需要引入以下指标:
-
QV 值(Quality Value): 衡量碱基层面的准确性。QV = -10 * log10(P_error)。例如,QV50 意味着每 10 万个碱基才有一个错误,这是 T2T 组装通常追求的标准。
-
BUSCO(Benchmarking Universal Single-Copy Orthologs): 衡量基因组的 完整性(Completeness)。它利用进化上高度保守的单拷贝直系同源基因作为“探针”,检查组装结果中包含了多少这些核心基因。如果 BUSCO 分数低(例如 < 90%),说明组装丢失了大量编码区信息,即便 N50 很高也是失败的。
-
k-mer 完整性: 比较组装结果的 k-mer 频谱与原始测序数据的 k-mer 频谱。理想情况下,组装结果应包含测序数据中出现的所有高频 k-mer,而不应包含测序数据中不存在的 k-mer(后者通常意味着组装错误)。
总结
从二代测序被迫使用破碎的 k-mer 图,到三代测序回归直观的 OLC 逻辑,再到 T2T 时代的多技术融合,基因组组装是算力、算法与数据质量三者之间的动态博弈。