在处理高通量组学数据(如转录组)时,我们往往会得到成百上千个“差异基因”。面对这份冗长的清单,仅关注单个基因很难把握生物学的全貌。富集分析(Enrichment Analysis)旨在将基因按照既定的生物学功能或通路(如 KEGG, GO)进行降维,帮助我们回答一个核心问题:这些发生变化的基因,作为一个整体,暗示了什么生物学过程的改变?
本文将梳理几种主流富集分析方法的演进逻辑。特别是各种方法在统计假设上的局限,以及其他方法是如何进一步做的,他们的使用场景等。
基本概念
前景基因 (Foreground):感兴趣的基因列表,例如实验组对比对照组筛选出的差异表达基因。 背景基因 (Background):实验中所有能被检测到且具有功能注释的基因集合。 基因集 (Gene Set):预定义的功能组合,例如“TCA循环”通路中包含的所有基因。
ORA 方法
过度富集分析 (Over-Representation Analysis, ORA) 是最直观的方法。它的核心逻辑是“数数”:如果某个通路在我们的差异基因列表中出现的频率,显著高于它在随机情况下(背景基因)出现的频率,我们就认为该通路被“富集”了。
数学上,这可以通过构建一个 列联表,并使用超几何分布或 Fisher 精确检验来计算概率。
下面演示如何利用 Python 进行 ORA 计算。
假设背景基因总数 ,某通路包含 个基因,我们筛选出的差异基因有 个,其中落入该通路的有 个。
import numpy as np
from scipy.stats import fisher_exact
# 列联表示例参数
N = 1000 # 背景基因总数
K = 50 # 通路基因数
M = 80 # 差异基因数
x = 10 # 差异基因中属于该通路的数目
# 构建2x2列联表
table = np.array([[x, M - x],
[K - x, N - K - (M - x)]])
print("2x2列联表:", table)
# Fisher 精确检验(右尾检验,测试富集)
oddsratio, p_value = fisher_exact(table, alternative='greater')
print(f"富集检验 p值 = {p_value:.3e}")背景基因的选择
进行 ORA 时,背景基因集的设定至关重要。背景应严格限定为实验中实际检测到且有注释的基因,而非物种的全基因组。如果背景过大(包含大量无法被检测到的基因),会人为稀释分母,导致 值虚低,产生假阳性结果。
ORA 的局限性在于它极其依赖阈值。你需要人为设定(例如 或 )来决定哪些是“差异基因”。这导致了两个问题:
- 信息丢失:一个 的基因被完全丢弃,而 的基因被保留,尽管它们在生物学上可能差别甚微。
- 忽略幅度:差异倍数是 10 倍的基因和 1.2 倍的基因在计数时被等同视之。
GSEA
为了解决阈值依赖问题,基因集富集分析 (GSEA) 引入了功能类打分 (Functional Class Scoring, FCS) 的概念。
GSEA 不需要预先筛选差异基因。它利用所有基因的表达数据,根据它们与表型的相关性(例如 Fold Change 或 t-statistic)进行排序。
算法从排序列表的顶端开始:遇到基因集内的基因就加分,遇到集外的基因就减分。最终得到的最大累积偏差就是富集得分 (Enrichment Score, ES)。
NOTE
GSEA 能够检测到那些单基因变化不显著、但作为一个整体协同变化的微弱信号。
以下代码模拟了 GSEA 的核心计算过程(ES 计算与置换检验):
import numpy as np
# 模拟数据
np.random.seed(1)
N = 50 # 基因总数
gene_set = set(range(10)) # 将前10个基因作为感兴趣基因集
# 为每个基因生成一个统计量,基因集成员均值抬高,非成员均值为0
stats = np.concatenate([np.random.normal(loc=2, scale=0.5, size=10),
np.random.normal(loc=0, scale=0.5, size=40)])
# 按统计量降序排序基因索引
ranked_indices = np.argsort(stats)[::-1]
# 计算富集得分 ES(Kolmogorov-Smirnov 累积和统计量)
N_set = len(gene_set)
N_total = len(ranked_indices)
hit_increment = 1.0 / N_set
miss_increment = 1.0 / (N_total - N_set)
running_sum = 0.0
ES = 0.0
for idx in ranked_indices:
if idx in gene_set:
running_sum += hit_increment
else:
running_sum -= miss_increment
if running_sum > ES:
ES = running_sum
# 置换检验估计 p值
n_perm = 1000
null_ES = []
for _ in range(n_perm):
# 随机打乱基因集成员身份
perm_set = set(np.random.choice(N_total, size=N_set, replace=False))
rs = 0.0
perm_ES = 0.0
for idx in ranked_indices: # 对同一排序进行走查
rs += hit_increment if idx in perm_set else -miss_increment
if rs > perm_ES:
perm_ES = rs
null_ES.append(perm_ES)
null_ES = np.array(null_ES)
p_value = (np.sum(null_ES >= ES) + 1) / (n_perm + 1)
print(f"模拟富集得分 ES = {ES:.3f}, 置换检验 p ≈ {p_value:.3f}")MsigDB
gene familiy overview
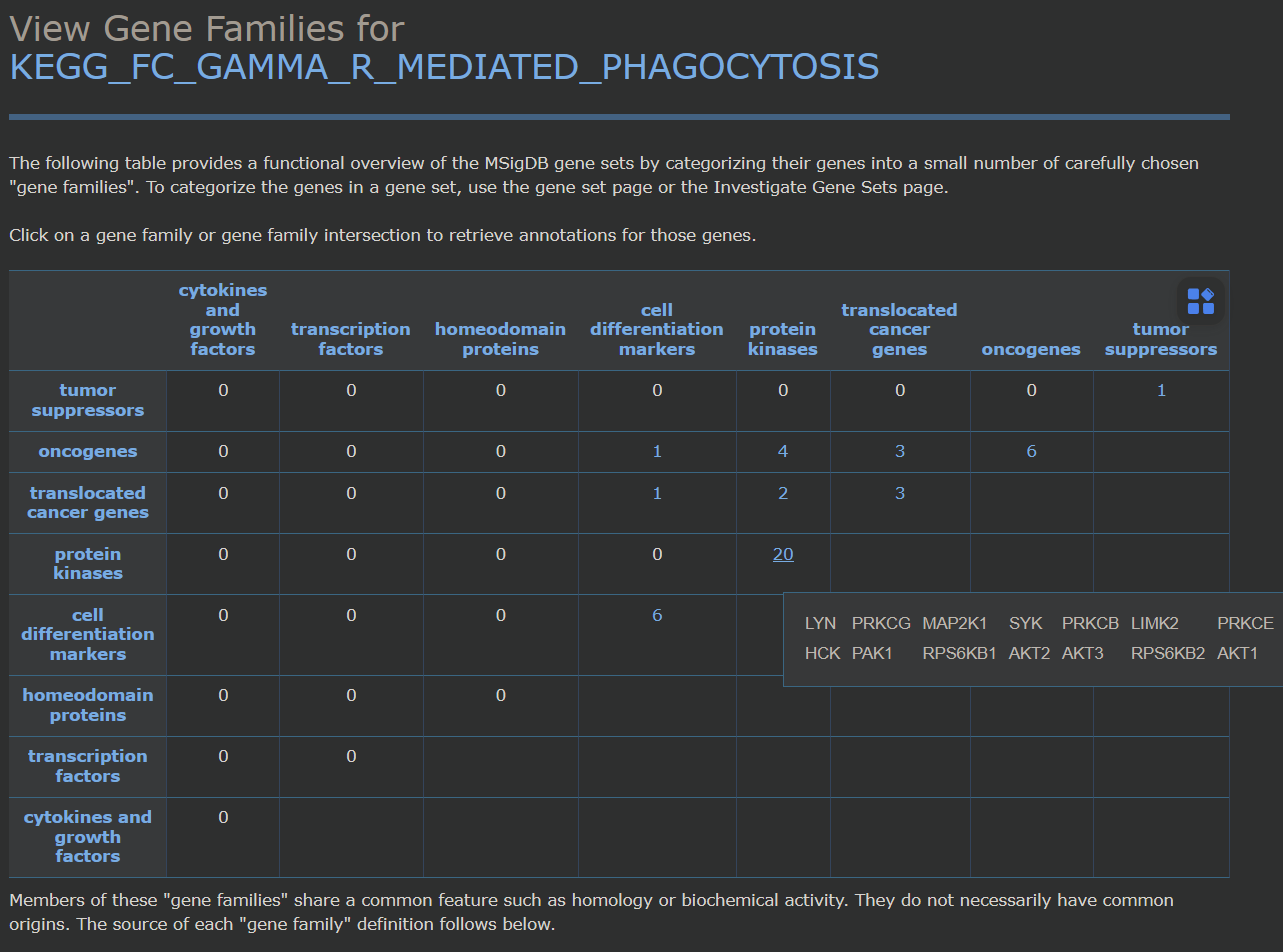
- 将gene按照其功能、蛋白家族进行划分
- 不同家族之间不是严格互斥:例如有的可能既是转录因子也是癌基因
- 二维更容易阅读,即使有的gene存在于多个familiy,这里只选用二维matrix
秩和检验如何用于富集分析?
GSEA 的 KS 统计量对分布两端的富集非常敏感。如果我们不关心这种“随机游走”的形态,而只关心基因集内的基因排名是否整体高于背景基因,可以使用更简单的非参数统计方法:Wilcoxon 秩和检验 (Rank-Sum Test)。
这种方法实质上是在比较两组数据的分布位置。如果基因集成员的统计量显著高于非成员,即可认为发生了富集。它计算速度快,且不依赖正态分布假设。
import numpy as np
from scipy.stats import ranksums
# 延续上一节模拟的数据 (stats 和 gene_set)
group1 = stats[list(gene_set)] # 基因集成员的统计量
group2 = stats[[i for i in range(N) if i not in gene_set]] # 其他基因的统计量
# Wilcoxon 秩和检验(假设备择:组1统计量大于组2)
stat, p_val = ranksums(group1, group2, alternative='greater')
print(f"秩和检验统计量 = {stat:.2f}, 单侧 p值 = {p_val:.4f}")
print("基因集成员平均统计量 = %.3f,其他基因平均统计量 = %.3f"
% (np.mean(group1), np.mean(group2)))GSEA 和秩和检验都属于竞争型 (Competitive) 检验,原假设是“该基因集并不比背景基因更相关”。
单细胞数据为何需要 AUCell?
上述方法主要面向群体水平(Bulk)的比较(如“治疗组 vs 对照组”)。但在单细胞测序(scRNA-seq)中,数据极其稀疏,且我们往往希望在单个细胞的层面上评估某通路的活跃程度,以便进行细胞分群或轨迹推断。
AUCell 方法应运而生。它基于排名,计算基因集成员占据该细胞高表达基因排名的“曲线下面积”(AUC)。如果基因集内的基因大量出现在该细胞表达量最高的前 5% 中,则 AUC 值高。
# 定义两个细胞的基因表达
expr_cell1 = {"G1": 8, "G2": 2, "G3": 7, "G4": 3, "G5": 1, "G6": 4} # 细胞1
expr_cell2 = {"G1": 1, "G2": 5, "G3": 0, "G4": 6, "G5": 4, "G6": 7} # 细胞2
gene_set = {"G1", "G3"} # 感兴趣基因集
# 计算单个细胞的基因集AUC得分
def auc_score(expr_values, gene_set):
genes_sorted = sorted(expr_values, key=expr_values.get, reverse=True)
N = len(genes_sorted)
k = len(gene_set)
ranks = {gene: rank+1 for rank, gene in enumerate(genes_sorted)}
# 计算恢复曲线的AUC:对每个基因集成员计算 (N - rank + 1)/(Nk) 并累加
auc = sum((N - ranks[g] + 1) / (N k) for g in gene_set if g in ranks)
return auc
auc1 = auc_score(expr_cell1, gene_set)
auc2 = auc_score(expr_cell2, gene_set)
print(f"Cell1 的基因集AUC得分: {auc1:.3f}")
print(f"Cell2 的基因集AUC得分: {auc2:.3f}")这种方法不依赖于细胞分组,能够直接揭示细胞间的异质性。
NOTE
competitive test的零假设是检验基因集不比背景更显著,self-contained test的零假设是检验基因集中没有基因发生变化。 ORA/GSEA/wilcox sum rank都是competitive test。
permuation和gene相关性破坏
sample label permutation不会影响样本内部gene之间的相关关系. gene label permuation则是则破坏了原先的相对关系。当实验样本量<7时, 将不得不使用gene set置换,这时会破坏gene相关性导致检验过于宽松,应该使用更严格阈值
基因集内的基因独立吗?
目前讨论的方法都有一个隐含的统计假设:基因集内的基因是独立的。
然而在生物学上,同一通路下的基因往往是共表达(Co-expressed)的。如果忽略这种相关性,会导致统计量的方差被严重低估,从而人为地放大显著性(假阳性膨胀)。简单来说,如果你有 50 个基因,它们完全共表达,那么你实际上只观测到了 1 个独立的信号,而不是 50 个。
CAMERA
CAMERA (Correlation Adjusted MEan RAnk) 方法引入了方差膨胀因子 (Variance Inflation Factor, VIF) 来校正基因相关性问题。
其中 是基因数, 是基因间的平均相关系数。即便 很小(如 0.05),只要 很大,VIF 也会显著大于 1,从而对检验统计量进行惩罚。
# 假设基因集大小和平均相关系数
k = 50
rho = 0.2
VIF = 1 + (k - 1) rho
# 比较独立假定和相关校正下,基因集统计量(如均值)的方差
var_independent = 1.0 / k # 独立时方差 ~ 1/k(假设每个基因统计量方差为1)
var_correlated = var_independent VIF # 考虑相关性的方差
print(f"基因数 m={k}, 平均相关 rho={rho:.2f}")
print(f"独立假定下统计量方差 ≈ {var_independent:.3f}")
print(f"相关校正后统计量方差 ≈ {var_correlated:.3f} (膨胀因子≈{VIF:.2f})")
print(f"折算的独立等效基因数 ≈ {k / VIF:.1f}")从输出可以看到,引入相关性后,有效样本量急剧下降。CAMERA 通过这种严谨的方差估计,让富集结果更加可靠。
SPIA
SPIA (Signaling Pathway Impact Analysis,信号通路影响分析) 直接考虑了不同gene在调控网络中的不同地位(上下游、FC大小,调控效果,)分别计算过表达、perturb两类pvalue,然后计算pvalue(global pvalue)。 perturb pvalue考虑了gene在网络中的位置,激活抑制类型,FC,效应如何在网络之内传播的。